BAC Clone Sequencing
Many researchers need perfectly accurate sequence of just a few BAC clones, from a few regions of interest.
Quick Strategic Overview with PacBio:
- Make High Quality NGS grade BAC DNA (minimizing host E. coli genomic DNA) – or you may provide us with ~10µg of ultra pure BAC DNA per clone.
- Make a high quality PacBio library (with 20Kb fragments and tags to allow combining several BAC’s in each PacBio run).
- Run the PAcBio reads through the HGAP DNA assembly pipeline to obtain as few contigs as possible.
- Genomic Content will impact assembly results. Our experience of typical results for 10 random BAC clones: 7 BAC’s will have a single contig (no gaps), 2 BAC’s will have 1 or 2 gaps and one BAC will have more than 3 gaps.
Contact us to get a BAC Clone Sequencing quote.
SEQUENCE THE MOST CHALLENGING BACS
Scientists often use bacterial artificial chromosome (BAC) sequencing to close gaps in existing genome assemblies, or as a method of targeted sequencing to fine-map regions of interest. BAC clones containing GC- or AT-rich regions or repeat structures can be challenging for short-read sequencing technologies.
With the long reads and uniform coverage of Single Molecule, Real-Time (SMRT) Sequencing, we can sequence and assemble finished BACs with high consensus accuracy – often into a single contig per BAC.
SMRT Sequencing on PacBio Systems gives you the ability to:
- Reconstruct medically and developmentally important structures within complex regions, such as the Y chromosome
- Finely map genes of interest with structural variation and haplotype information for evolutionary and agricultural studies
- Generate platinum-level genome assemblies
- Resolve historically difficult-to-sequence regions in draft genomes
- Pool dozens of BACs in a single SMRT Cell for efficient and cost-effective sequencing
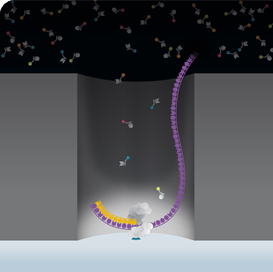
Workflow: Using BACs to close coverage of the most complex regions
 |
Library Preparation
|
|
|
SMRT Sequencing on PacBio Systems
|
 |
Data Analysis with SMRT Analysis
|

Contact us to get a BAC Clone Sequencing quote.