Below you will find some common problems associated with DNA sequencing as well as the possible causes and solutions for these problems. A picture of the sequence traces is presented above the information describing the problem, how to identify the problem, the cause, and the potential solution for the problem. Other problems can occur with sequence data, but the ones presented below are those seen most commonly.
Compression Sequence
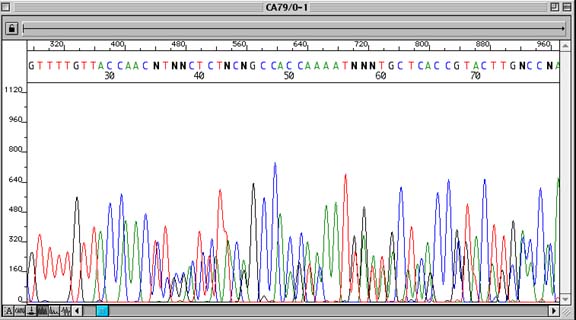
Problem:
Sequence appears as multiple overlying traces, not always throughout the entire sequence
How to Identify:
Sequence data begins to show multiple overlapping traces after a point in the sequence, although the sequencing signal remains strong. A compression phenomenon and contaminated DNA prep can look similar, though the contaminated prep will typically show overlapping traces immediately after the insert cloning site.
Cause:
DNA fragments of different size with the same electrophoretic mobility, ie fragments that migrate on top of each other during electrophoresis. This phenomenon is thought to be caused by regions of secondary structure within the template DNA, which are often, but not always, found in regions with a high G/C or high A/T content.
Solution:
- Add DMSO to a final 5% (v/v) concentration to the sequencing reaction.
- Incubate the reaction at 96C for 10 minutes before cycle sequencing.
- Double all reaction components and incubate at 98C for 10 minutes before cycling. (Note: Reaction components are doubled to account for the amount of enzyme that is inactivated by the higher denaturation temperature. Do not use this solution when using the Sequencing Reagent Dilution Buffers)
- Linearize plasmids with a restriction enzyme.
- If the sample is a PCR product, try amplifying the DNA with substitution of 7-deaza-dGTP for 75% of the dGTP in the PCR, and then sequence the PCR product.
- Choose a new primer close to the compression site which can help avoid the effects of the secondary structure.
Contaminated Plasmid DNA Preparation
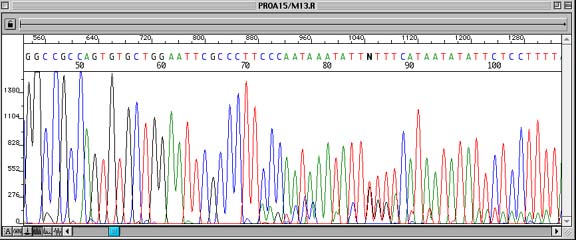
Problem:
Contaminated plasmid DNA preparation leads to overlapping peaks in the sequence data.
How To Identify:
Sequence data is clear out to the end of the insert cloning site, at which point peaks underneath other peaks can often be seen. In the worst case, the peaks will obscure each other, making the entire sequence unreadable.
Cause:
Picking more than one clone for a single plasmid preparation, or picking a single colony that contains two or more plasmids, each with different inserts.
Solution:
- Carefully pick a single colony from your plate into your growth medium in a clean environment.
- Verify that your single colony contains your insert of interest after DNA preparation by restriction digest and agarose gel analysis.
High Background/Noisy Sequencing Signal
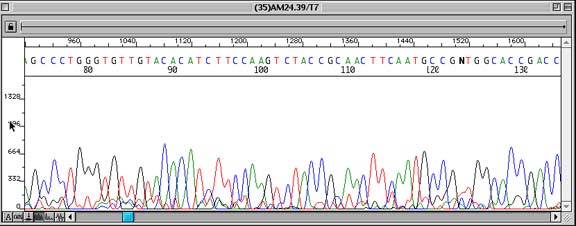
Problem:
Low/Poor Signal-to-noise ratio in sequence data causing unreadable sequence data at points in the sequence.
How to Identify:
Observation of small peaks underneath all peaks in a sequence read. Low signal can be confirmed by checking the ‘information view’ (click on the ‘A:’ radio key seen at the bottom left of an EditView chromatogram trace) and verifying that Average Signal Intensity is less than 100 relative fluorescent units (rfu). In this picture, the average sequencing signal for each base was as follows: G (44), A (50), T (20), C (47). Note: It is also possible that ‘double peaks’ throughout a sequence are suggestive of the primer binding to more than one site on the DNA template. However, the fluorescent intensity numbers would suggest that the above results were caused by low signal.
Cause:
- Too little DNA template is added to sequencing reaction.
- Primer binding to the template is not very efficient.
- Not enough Big Dye Terminator Mix was added to the sequencing reaction
- The sequencing reaction was not very efficient, as is sometimes the case with BAC end- or direct BAC sequencing.
- DNA template was not purified well enough prior to the sequencing reaction and is contaminated.
Solution:
- For (1), increase the amount of template added to the reaction.
- For (2), increase the amount of primer in the reaction, or if possible, design a new primer.
- With (3), increase the amount of BDT mix added to the reaction.
- For (4), you can shear BAC DNA by vortexing it at maximum rpm for 1 minute or passing it through a 28 gauge sterile needle, digest it with a restriction enzyme prior to sequencing, and denaturing the BAC DNA prior to adding to the sequencing reaction (98C for 10 min).
- Prepare the DNA again using fresh reagents and careful techniques.
Gradual Early Termination of Sequencing Signal
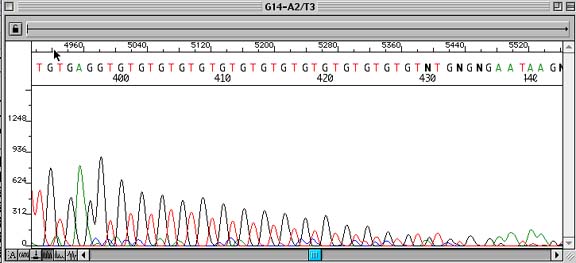
Problem:
Premature termination of sequence due to repetitive structure(s) of DNA.
How to identify:
Repetitive stretches of DNA sequence (usually mono- or dinucleotide repeats) that show strong sequencing signals at the beginning of the repeat and then gradually taper off to an unreadable signal.
Cause:
Various ideas have been put forth on this subject: (1) Slippage of the DNA polymerase on the template strand during elongation (2) Formation of secondary structure due to the repeat. (3) Depletion of dNTPs.
Solution:
Sequence the template from the other side of the repeat in the reverse direction to obtain sequence on both sides of the repeat. Be aware that it will be difficult to determine the precise length of the repeat if neither sequencing reaction manages to span the entire length of the repeat. Although it may not often be practical, using different sequencing chemistries may improve results as well.
N-1, N-2, N-3… Priming Sequencing Trace
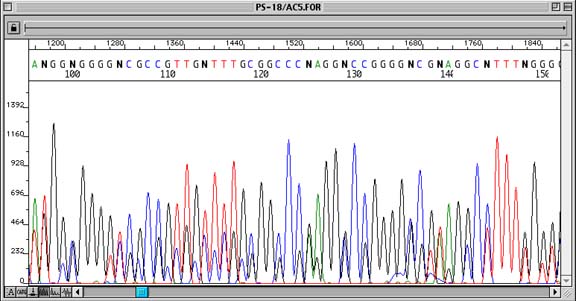
Problem:
Sequence shows ‘double peaks’ throughout, with the second (generally smaller) peak being the same base as that of the true base immediately to the right of it.
How to identify:
Although an n-1 primer problem may look like compression or priming of two different regions of DNA template, it can be readily identified by each position containing the true peak and the peak immediately to the right of it.
Cause:
- Poor purification of the oligonucleotide during primer synthesis which leaves a certain percentage of n-1mers in the final product. When the DNA template is sequenced, this percentage of n-1mers will prime the DNA template, causing some of the sequence to be 1 bp shorter than it should be. Primers that have begun to degrade will also do so from the 3′ end, causing a proportion of the original sequencing primer to become n-1.
- The primer has started to degrade from the 3′ end so that a portion of the sequences will have a sequencing ladder that is one basepair longer than a non-degraded primer.
- Poor primer binding site on the template.
Solution:
Cartridge purification of oligonucleotides and even HPLC purification will help insure that n-1 priming of sequence doesn’t occur. In both cases, re-synthesis of the oligonucleotide is necessary.
No Readable Sequence Data
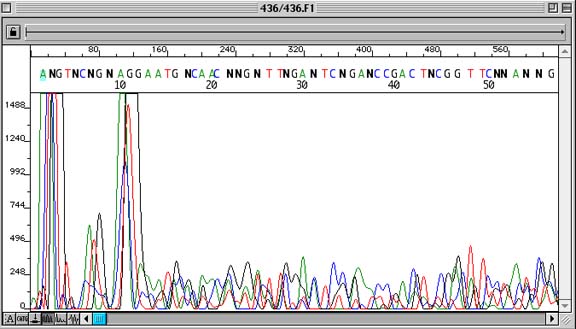
Problem:
Lack of sequence data
How to identify:
Sequencing trace is non-uniform. Unincorporated Dye Terminator Peaks at the beginning of the sequence are out-of-scale. Basecalls contain many ambiguous bases (‘N’).
Cause:
- Primer not annealing to template (no priming site).
- No DNA template present in sequencing reaction.
- Incorrect cycle sequencing conditions.
- Template not denaturing during cycle sequencing.
- DNA template is of poor quality.
Solution:
- For (1), design a new primer which is free of hairpins, free of duplexes, and has a Tm greater than 55C.
- For (2), it is important to verify DNA concentrations before addition to the sequencing reaction in order to insure that the optimum amount of template is being added to the reaction.
- For (3), verify that your cycle sequencing conditions are correct, and if necessary, modify them.
- For (4), either incubate your sequencing reaction at 96C for 10 min prior to cycle sequencing or incubate the template only at 98C for 10 minutes before addition to the sequencing reaction.
Secondary Structure
Problem:
Sharp drop or premature termination of sequencing signal during the sequencing run.
How to identify:
Sequencing signal drops off sharply or terminates before the end of the sequencing run (termination of sequencing signal also occurs with PCR products, depending on how long the PCR fragment is).
Cause:
Secondary structure in the template can cause various anomalies that result in inefficient chain elongation after the region of secondary structure.
Solution: Many of the same techniques used to eliminate compression can be helpful in obtaining good sequence data past regions of secondary structure. Although not always practical, changing sequencing chemistries can produce better results as well.
At Amplicon Express, we use strict protocols and internal quality controls to insure our reactions are as consistent as possible. Most often when there is trouble with the sequencing reaction, the cause lies with the DNA template itself, making it very important that the materials we sequence are of the highest quality possible. If you have further questions or need technical support, please contact us.
For a more complete reference, see DNASequencingGuide.